
โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลัง
| โรคกล้ามเนื้อเสียการประสานงาน จากสมองน้อยและไขสันหลัง (Spinocerebellar ataxia) | |
|---|---|
| ชื่ออื่น | Spinocerebellar atrophy or Spinocerebellar degeneration |
 | |
| ก้อนสีม่วงคือสมองน้อย (cerebellum) ของมนุษย์ | |
| สาขาวิชา | ประสาทวิทยา |
โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลัง (อังกฤษ: spinocerebellar ataxia; spinocerebellar degeneration) เป็นโรคทางพันธุกรรมโรคหนึ่งซึ่งปัจจุบันยังไม่มีหนทางเยียวยา ผู้ป่วยจะสูญเสียความสามารถในการเคลื่อนไหวทางกายภาพไปโดยช้าจนกระทั่งหมดสิ้น โรคนี้มีหลายชนิด ซึ่งในแต่ละชนิดอาการแสดง อายุของผู้ป่วยที่เริ่มเป็นโรค และลักษณะการถ่ายทอดทางพันธุกรรมจะแตกต่างกันไปขึ้นอยู่กับตำแหน่งของยีนบนโครโมโซมของผู้ป่วยที่ได้รับผลกระทบ [1]
ภาพรวมของโรค[แก้]
โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังนั้นถ่ายทอดทางพันธุกรรมซึ่งทำให้สมองน้อย (cerebellum) ไขสันหลัง (spinal cord) และระบบประสาทนอกส่วนกลาง (peripheral nervous system) ซึ่งอยู่บริเวณไขสันหลังของผู้ป่วยทำงานผิดปรกติเนื่องจากมีการฝ่อลีบลง โรคนี้จะปรากฏโดยช้าและรุนแรงขึ้นเรื่อย ๆ อย่างไม่สามารถยับยั้งได้ [1]
สำหรับสมองน้อยของมนุษย์นั้นทำหน้าที่ร่วมกับระบบประสาททั้งส่วนกลาง (central nervous system) และส่วนปลาย (peripheral nervous system) ในการควบคุมการทรงตัวและการเคลื่อนไหวของมนุษย์ โดยอาศัยไขสันหลังในการส่งผ่านกระแสประสาท
ความชุกของโรค[แก้]
โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังพบได้น้อยมากในโลกนี้[2] โดยมีอัตราการเกิดโรคประมาณสิบหกคนต่อหนึ่งแสนคน [3]
อาการของโรค[แก้]
อาการของโรคนี้มักเริ่มปรากฏตั้งแต่ผู้ป่วยยังอยู่ในวัยก่อนวัยเริ่มเจริญพันธุ์ (puberty) อย่างไรก็ดี โรคนี้บางประเภทก็ปรากฏในวัยผู้ใหญ่ (adult) เลยก็ได้
ผู้ป่วยเป็นโรคนี้จะสูญเสียความสามารถในการควบคุมการเคลื่อนไหวทางกายภาพของตน โดยเกิดภาวะสูญเสียการประสานกันของท่าเดิน (unsteady gait) มีอาการพูดไม่เป็นความ (dysarthria) และอาการตากระตุก (nystagmus) นอกจากนี้ ผู้ป่วยยังอาจมีอาการอื่น ๆ ร่วมด้วยนอกเหนือจากดังกล่าว เช่น อาการสั่น (tremor) ภาวะซึมเศร้า (depression) ภาวะหดเกร็งของกล้ามเนื้อ (spasticity) และโรคนอนไม่หลับ (sleep disorders) อนึ่ง การค้นคว้าอื่น ๆ ยังระบุว่า อาจมีโรคดังต่อไปนี้แทรกซ้อนขึ้นมาในระหว่างการป่วยได้ เช่น ภาวะกระดูกสันหลังโกงคด (kyphoscoliosis) ภาวะนิ้วเท้างุ้ม (hammer toe) ภาวะส่วนโค้งเท้าสูงขึ้น (high arches) หรือโรคหัวใจ (heart disease)
อาการแสดง อายุของผู้ป่วยที่เริ่มเป็นโรค และลักษณะการถ่ายทอดทางพันธุกรรมจะแตกต่างกันไป ขึ้นอยู่กับตำแหน่งของยีนบนโครโมโซมที่ได้รับผลกระทบของผู้ป่วย อย่างไรก็ดี แม้ความสามารถในการเคลื่อนไหวทางกายภาพของผู้ป่วยจะเสื่อมสภาพลงตามลำดับและสูญเสียไปในที่สุด แต่ระบบจิตใจและความรู้สึกนึกคิดของผู้ป่วยยังคงปรกติ
สาเหตุ[แก้]
โรคนี้เกิดขึ้นจากความผิดปรกติในการผลิตซ้ำของโพลีกลูตาไมน์ ไตรนิวคลีโอไทด์ (Trinucleotide repeat disorders, trinucleotide repeat expansion disorders, triplet repeat expansion disorders หรือ codon reiteration disorders) อันเป็นความผิดปรกติทางพันธุกรรมเพราะนิวคลีโอไทด์ผลิตตนเองซ้ำ ๆ มากกว่าปรกติและเกินความต้องการของร่างกาย หากการผลิตดังกล่าวมีขึ้นเป็นเวลานานอาจทำให้เกิดโรคดังกล่าวได้เร็วขึ้น และจะมีการดำเนินโรคและอาการทางคลินิกมากขึ้น
ชนิดของโรค[แก้]
โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังสามารถจำแนกชนิดได้โดยอาศัยรูปแบบการถ่ายทอดทางพันธุกรรมและยีนในโครโมโซมที่เกิดความผิดปกติ โดย โรคนี้สามารถถ่ายทอดได้ทั้งทางพันธุกรรมลักษณะเด่น (autosomal dominant) และลักษณะด้อย (autosomal recessive)
การถ่ายทอดทางพันธุกรรมลักษณะเด่น[แก้]
สำหรับโรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังที่ถ่ายทอดทางพันธุกรรมลักษณะเด่นนั้น เมื่อบุคคลได้รับยีนผิดปรกติเพียงยีนหนึ่งจากบิดาหรือมารดาก็มีโอกาสป่วยเป็นโรคนี้ได้ถึงร้อยละห้าสิบ เช่นเดียวกับบุตรของผู้ป่วยซึ่งก็มีโอกาสป่วยเป็นโรคนี้ได้ถึงร้อยละห้าสิบเช่นเดียวกัน[4]
ประเภทของโรค[แก้]
โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังยังแบ่งได้อีกหลายประเภทตามแต่ตำแหน่งของยีนบนโครโมโซมที่ได้รับผลกระทบ ทั้งนี้ ดังรายละเอียดในตารางต่อไปนี้
| ชื่อชนิด | ช่วงเวลาตั้งต้นของโรคโดยเฉลี่ย | ความยาวนานของโรค | อาการเด่นชัดของผู้ป่วย | แหล่งที่พบมาก | อาการในโครงสร้างทางพันธุกรรม |
|---|---|---|---|---|---|
| เอสซีเอ-1 (อะทาซิน-1 เก็บถาวร 2012-02-21 ที่ เวย์แบ็กแมชชีน) [5] | สี่สิบปี (น้อยกว่าสิบปีหรือ อาจเกินกว่าหกสิบปี) |
สิบห้าปี (สิบถึงยี่สิบแปดปี) |
- hypermetric saccades - ภาวะสูญเสียเซลล์ประสาทสั่งการส่วนบน (loss of upper motor neuron) - slow saccades |
- โครโมโซม 6-พี (อะทาซิน 1) และ - การผลิตซ้ำของโครงสร้างทางพันธุกรรมอะมิโนกลูตาไมน์ (CAG repeated sequence) | |
| เอสซีเอ-2 (อะทาซิน-2 เก็บถาวร 2012-02-21 ที่ เวย์แบ็กแมชชีน) [6] | สามสิบปีถึงสี่สิบปี (น้อยกว่าสิบปีหรือ อาจเกินกว่าหกสิบปี) |
สิบปี (หนึ่งปีถึงสามสิบปี) |
ภาวะสูญเสียรีเฟล็กซ์ (areflexia) | คิวบา | - โครโมโซม 12-คิว และ -การผลิตซ้ำของโครงสร้างทางพันธุกรรมอะมิโนกลูตาไมน์ (CAG repeated sequence) |
| - เอสซีเอ-3 (อะทาซิน-3 เก็บถาวร 2012-02-21 ที่ เวย์แบ็กแมชชีน) หรือ - โรคมาชาโด-โจเซฟ (Machado-Joseph disease) [7] [8] |
สี่สิบปี (สิบปีถึงเจ็ดสิบปี) |
สิบปี (หนึ่งปีถึงยี่สิบปี) |
- อาการตากระตุกขณะเพ่งมอง (Gaze-evoked nystagmus) - ภาวะสูญเสียเซลล์ประสาทสั่งการส่วนบน (loss of upper motor neuron) - slow saccades |
หมู่เกาะอซอส์ (Azores) โปรตุเกส |
- โครโมโซม 14-คิว และ - การผลิตซ้ำของโครงสร้างทางพันธุกรรมอะมิโนกลูตาไมน์ (CAG repeated sequence) |
| เอสซีเอ-4 (พีแอลอีเคเอชจี-4[ลิงก์เสีย]) | สี่สิบปีถึงเจ็ดสิบปี (สิบเก้าปีถึงเจ็ดสิบสองปี) |
หลายสิบปี | ภาวะสูญเสียรีเฟล็กซ์ (areflexia) | โครโมโซม 16-คิว | |
| เอสซีเอ-5 (เอสพีทีบีเอ็น-2[ลิงก์เสีย]) | สามสิบปีถึงสี่สิบปี (สิบปีถึงหกสิบแปดปี) |
ยี่สิบห้าปีขึ้นไป | pure cerebellar | โครโมโซม 11 | |
| เอสซีเอ-6 (ซีเอซีเอ็นเอวันเอ เก็บถาวร 2011-09-19 ที่ เวย์แบ็กแมชชีน) [9] | ห้าสิบปีถึงหกสิบปี (สิบเก้าปีถึงเจ็ดสิบเอ็ดปี) |
ยี่สิบห้าปีขึ้นไป | - อาการตากระตุกลง (downbeating nystagmus) - positional vertigo (อาการแสดงของชนิดนี้มักเกิดขึ้นเป็นครั้งแรก อย่างช้าเมื่ออายุ 65 ปี) |
- การผลิตซ้ำของกรดอะมิโนกลูตาไมน์ (CAG repeated sequence) - โครโมโซม 19-พี และ - ยีนช่องแคลเซียม (calcium channel gene) | |
| เอสซีเอ-7 (อะทาซิน-7 เก็บถาวร 2012-02-21 ที่ เวย์แบ็กแมชชีน) [10] | สามสิบปีถึงสี่สิบปี (ครึ่งปีถึงหกสิบปี) |
ยี่สิบปี (หนึ่งปีถึงสี่สิบห้าปี; การเกิดโรคครั้งแรกสัมพันธ์กับระยะเวลาของโรคที่สั้น) |
- การเสื่อมที่จุดภาพชัด (macular degeneration) - ภาวะสูญเสียเซลล์ประสาทสั่งการส่วนบน (loss of upper motor neuron) - slow saccades |
- โครโมโซม 3-พี (อะทาซิน 7) และ - การผลิตซ้ำของกรดอะมิโนกลูตาไมน์ (CAG repeated sequence) | |
| เอสซีเอ-8 (ไอโอเอสซีเอ[ลิงก์เสีย]) [11] | สามสิบเก้าปี (สิบแปดปีถึงหกสิบห้าปี) |
ช่วงชีวิตปรกติ | อาการตากระตุกในแนวราบ (horizontal nystagmus) | - โครโมโซม 13-คิว และ - การผลิตซ้ำของลำดับพันธุกรรมซีทีจี (CTG repeated sequence) | |
| เอสซีเอ-10 (อะทาซิน-10[ลิงก์เสีย]) [12] | สามสิบหกปี | เก้าปี | กล้ามเนื้อเสียการประสานงาน (ataxia) และการชัก (seizure) | เม็กซิโก | - โครโมโซม 22-คิว และ - การผลิตซ้ำของโครงสร้างทางพันธุกรรมเพ็นทานิวคลีโอไทด์ (pentanucleotide repeated sequence) |
| เอสซีเอ-11[ลิงก์เสีย] | สามสิบปี (สิบห้าปีถึงเจ็ดสิบปี) |
ช่วงชีวิตปรกติ | ภาวะอ่อนแอ (mild) [แต่ยังสามารถเดินได้ด้วยตนเอง (ambulatory)] |
โครโมโซม 15-คิว | |
| เอสซีเอ-12 (พีพีพีทูอาร์ทูบี[ลิงก์เสีย]) [13] | สามสิบสามปี (แปดปีถึงห้าสิบห้าปี) |
- อาการสั่นของศีรษะและมือ (head and hand tremor) และ -ภาวะเสียการเคลื่อนไหว (akinesia) |
- โครโมโซม 5-คิว และ - การผลิตซ้ำของกรดอะมิโนกลูตาไมน์ (CAG repeated sequence) | ||
| เอสซีเอ13 | ขึ้นอยู่กับการกลายพันธุ์ | ขึ้นอยู่กับพันธุกรรมเคเอ็นซีเอ็น 3 (KCNC3, พันธุกรรมชนิดหนึ่ง) |
ภาวะปัญญาอ่อน (mental retardation) | โครโมโซม 19-คิว | |
| เอสซีเอ-14 (พีอาร์เคซีจี เก็บถาวร 2008-05-29 ที่ เวย์แบ็กแมชชีน) [14] | ยี่สิบแปดปี (สิบสองปีถึงสี่สิบสองปี) |
หลายสิบปี (หนึ่งปีถึงสามสิบปี) |
ภาวะกล้ามเนื้อกระตุกรัว (myoclonus) | โครโมโซม 19-คิว | |
| เอสซีเอ-16 | สามสิบเก้าปี (ยี่สิบปีถึงหกสิบหกปี) |
หนึ่งปีถึงสี่สิบปี | อาการสั่นของศีรษะและมือ (head and hand tremor) | โครโมโซม 8-คิว | |
| เอสซีเอ-17 (ทีบีพี เก็บถาวร 2008-05-20 ที่ เวย์แบ็กแมชชีน) | - การผลิตซ้ำของโครงสร้างทางพันธุกรรมอะมิโนกลูตาไมน์ (CAG repeated sequence) และ - โครโมโซม 6-คิว (พันธะโปรตีนโกลด์เบิร์ก-ฮอกเนสบ็อกซ์, TATA-binding protein) | ||||
| เอสซีเอ-19[ลิงก์เสีย] และ เอสซีเอ-22[ลิงก์เสีย] |
- อาการสมองน้อยอ่อน (Mild cerebellar syndrome) และ - อาการพูดไม่เป็นความ (dysarthria) |
||||
| เอสซีเอ-25[ลิงก์เสีย] | หนึ่งปีครึ่งถึงสามสิบเก้าปี | ไม่ทราบ | - กล้ามเนื้อเสียการประสานงานเหตุประสาทพิการ - การอาเจียน และ - ภาวะเจ็บปวดในทางเดินอาหาร (gastrointestinal tract pain) |
โครโมโซม 2-พี |
หมายเหตุ
- "ซีเอซีเอ็นเอวันเอ" (CACNA1A) หมายถึง ช่องแคลเซียมอาศัยศักย์ไฟฟ้าประเภทพี/คิว หน่วยย่อยแอลฟา 1-เอ (calcium channel, voltage-dependent, P/Q type, alpha 1-A subunit)
- "ทีบีพี" (TBP) หมายถึง พันธะโปรตีนโกลด์เบิร์ก-ฮอกเนสบ็อกซ์ (TATA box binding protein)
- "พีพีพีทูอาร์ทูบี" (PPP2R2B) หมายถึง โปรตีนฟอสฟาเทส 2 ส่วนควบคุมย่อยบี บีทาไอโซฟอร์ม (protein phosphatase 2, regulatory subunit B, beta isoform)
- "พีอาร์เคซีจี" (PRKCG) หมายถึง โปรตีนไคเนส-ซี, แกมมา (protein kinase-C, gamma)
- "พีแอลอีเคเอชจี" (PLEKHG) หมายถึง แพล็กสทรินฮอมอโลยี-ตระกูลจี (pleckstrin homology-family G)
- "อะทาซิน" (ataxin) หมายถึง กล้ามเนื้อเสียการประสานงาน
- "เอสซีเอ" (SCA) หมายถึง โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลัง
- "เอสพีทีบีเอ็น" (SPTBN) หมายถึง สเปกตริน บีทา นอน-อิริโทรไซคลิก (spectrin, beta, non-erythrocytic)
- "ไอโอเอสซีเอ" (IOSCA) หมายถึง โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังซึ่งเริ่มเกิดในเด็ก (infantile onset spinocerebellar ataxia)
- "ความยาวนานของโรค" หมายถึง ระยะเวลาที่ผู้ป่วยจะมีชีวิตอยู่
การถ่ายทอดทางพันธุกรรมลักษณะด้อย[แก้]
โรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังที่ถ่ายทอดทางพันธุกรรมลักษณะด้อยนั้น อาศัยยีนผิดปกติทั้งจากบิดาและมารดาจึงจะก่อโรคนี้ โดยมีโอกาสเกิดเพียงแค่ร้อยละยี่สิบห้าของการตั้งครรภ์หนึ่งครั้ง ชื่อของชนิดจะแตกต่างกันตามแต่ตำแหน่งของยีนบนโครโมโซมที่ได้รับผลกระทบ เช่น
- โรคกล้ามเนื้อเสียการประสานงานแบบเฟรดริก (Friedreich ataxia)
- โรคกล้ามเนื้อเสียการประสานงานและหลอดเลือดฝอยพอง (Ataxia-telangiectasia)
- โรคกล้ามเนื้อเสียการประสานงานร่วมกับการขาดวิตามินอี (Ataxia with vitamin E deficiency)
- โรคกล้ามเนื้อเสียการประสานงานร่วมกับการเสียการทำงานของประสาทกล้ามเนื้อตา (ataxia with oculomotor apraxia; AOA) และ
- โรคกล้ามเนื้อเสียการประสานงานชนิดกล้ามเนื้อเกร็ง (Spastic ataxia)
-
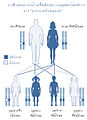 การถ่ายทอดโรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังแบบลักษณะเด่น
การถ่ายทอดโรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังแบบลักษณะเด่น -
 การถ่ายทอดโรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังแบบลักษณะด้อย
การถ่ายทอดโรคกล้ามเนื้อเสียการประสานงานจากสมองน้อยและไขสันหลังแบบลักษณะด้อย


การวินิจฉัยโรค[แก้]
การวินิจฉัยโรคนี้ต้องอาศัยความรู้ทางพันธุศาสตร์และประสาทวิทยาอย่างมาก เพราะอาจสับสนกับโรคทางระบบประสาท (neurological condition) อื่น ๆ ได้ เป็นต้นว่า โรคปลอกประสาทเสื่อมแข็ง (multiple sclerosis)
วิธีหนึ่งในการวินิจฉัยโรคนี้ได้แก่การใช้ เครื่องเอกซเรย์คอมพิวเตอร์ (CT Scan) หรือ เครื่องฉายภาพด้วยเรโซแนนซ์แม่เหล็ก (MRI) ในการตรวจวิเคราะห์สมอง หากป่วยเป็นโรคนี้จริงจะพบว่าสมองน้อยของผู้ป่วยฝ่อลงอย่างเห็นได้ชัด
การวินิจฉัยที่แม่นยำที่สุดได้แก่การตรวจโครงสร้างทางพันธุกรรมของผู้ป่วย วิธีนี้ยังสามารถใช้ตรวจวิเคราะห์เด็กว่ามีความเสี่ยงที่จะเป็นโรคทางพันธุกรรมนี้หรือไม่ด้วย
การบำบัดรักษาและการพยากรณ์โรค[แก้]
ปัจจุบันยังไม่มีวิธีรักษาผู้ป่วยเป็นโรคนี้ และไม่มีวิธีฟื้นฟูร่างกายผู้ป่วยให้กลับเป็นปกติเช่นเดิมได้ แต่ยังสามารถบรรเทาความรุนแรงของอาการได้ โดยอาศัยการทำกายภาพบำบัดและรักษาตามอาการที่เกิดขึ้น
การบรรเทาความรุนแรงของอาการต่าง ๆ นั้น ได้แก่ การใช้อุปกรณ์ภายนอกช่วยเหลือผู้ป่วยเพื่อให้สามารถดำรงชีวิตได้ด้วยตนเองมากที่สุดเท่าที่กระทำได้ เช่น ไม้เท้า (cane) ไม้ยันรักแร้ (crutch) เครื่องช่วยเดิน (walker) หรือเก้าอี้รถเข็น (wheelchair) ช่วยในการเคลื่อนไหว กับทั้งอุปกรณ์ช่วยเขียน ช่วยป้อนอาหาร และช่วยดูแลผู้ป่วยในกรณีที่สูญเสียความสามารถในการเคลื่อนไหวของมือ ตลอดจนอุปกรณ์ช่วยติดต่อสื่อสารในกรณีที่สูญเสียความสามารถของขากรรไกรล่างด้วย
ตัวอย่างผู้ป่วย[แก้]
- นายกลิน วอร์สนิป (Glyn Worsnip) (2 กันยายน 2481—7 มิถุนายน 2539) : ผู้ประกาศข่าวชาวบริติชแห่งสถานีโทรทัศน์บีบีซีซึ่งป่วยเป็นโรคนี้ชนิดที่ 19 (เอสซีเอ-19) และมีอาการพูดไม่เป็นความ (dysarthria) ในระหว่างประกาศข่าว ทำให้ถูกผู้ชมร้องเรียนต่อสถานีและถูกไล่ออกตามลำดับ นายกลินได้จัดสร้างรายการโทรทัศน์ชื่อ "อะโลนวอยซ์" (A Lone Voice) เกี่ยวกับโรคนี้ออกอากาศทางบีบีซี
- นางสาวอะยะ คิโต (Aya Kitō) (19 กรกฎาคม 2505—23 พฤษภาคม 2531) : เด็กสาวชาวญี่ปุ่นซึ่งเขียนบันทึกประจำวันเกี่ยวกับประสบการณ์การป่วยเป็นโรคนี้ของเธอ ภายหลังจากการตายของอะยะ บันทึกดังกล่าวได้รับการพิมพ์เผยแพร่ชื่อว่า "อิชิริตโตะรุโนะนะมิดะ" (Ichi Rittoru no Namida, น้ำตาหนึ่งลิตร) และได้รับการดัดแปลงเป็นละครโทรทัศน์ชื่อเดียวกัน ซึ่งละครเรื่องดังกล่าวได้ออกฉายทางทีวีไทย ชื่อไทยว่า "บันทึกน้ำตาหนึ่งลิตร" ตั้งแต่เดือนพฤษภาคม 2551
- ครอบครัวอูลาส (Ulas Family) : เป็นครอบครัวชาวตุรกีประกอบไปด้วยสมาชิกสิบเก้าคน สมาชิกห้าคนป่วยเป็นโรคนี้ทำให้สูญเสียความสามารถในการเดิน พวกเขาจึงเดินในอาการคลานด้วยเข่าและมือทั้งสี่ข้าง สถานีโทรทัศน์บีบีซีได้จัดทำสารคดีชื่อ "เดอะแฟมิลีแดตวอกส์ออนออลโฟส์" (The Family That Walks On All Fours) เกี่ยวกับชะตาชีวิตของครอบครัวนี้ ออกอากาศในสหราชอาณาจักรเมื่อ พ.ศ. 2549
อ้างอิง[แก้]
- ↑ 1.0 1.1 Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th ed. Philadelphia, Elsevier Saunders: 2005.
- ↑ Bangkokhealth.com. (2550, 30 พฤษภาคม). Spinocerebellar degeneration. [ออนไลน์]. เข้าถึงได้จาก: http://www.1719.net/consult_htdoc/Question.asp?GID=73359[ลิงก์เสีย]. (7 พฤษภาคม 2551).
- ↑ Sasaki et al. 2003 [1][ลิงก์เสีย]
- ↑ กรมสุขภาพจิต. (2548, 17 สิงหาคม). ภาวะเดินเซหรือ Ataxia คืออะไร. [ออนไลน์]. เข้าถึงได้จาก: http://www.dmh.go.th/sty_libnews/news/view.asp?id=2479 เก็บถาวร 2008-05-14 ที่ เวย์แบ็กแมชชีน. (7 พฤษภาคม 2551).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA1 [Online]. Available: http://www.geneclinics.org/profiles/sca1 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA2 [Online]. Available: http://www.geneclinics.org/profiles/sca2 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA3 [Online]. Available: http://www.geneclinics.org/profiles/sca3 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
- ↑ US's National Institute of Neurological Disorders and Stroke. (n.p.). Machado-joseph. (Online). Available: http://www.ninds.nih.gov/disorders/machado_joseph[ลิงก์เสีย]. (6 May 2008).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA6 [Online]. Available: http://www.geneclinics.org/profiles/sca6 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA7 [Online]. Available: http://www.geneclinics.org/profiles/sca7 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA8 [Online]. Available: http://www.geneclinics.org/profiles/sca8 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA10 [Online]. Available: http://www.geneclinics.org/profiles/sca10 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA12 [Online]. Available: http://www.geneclinics.org/profiles/sca12 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
- ↑ University of Washington’s National Center for Biotechnology Information. (n.p.) SCA14 [Online]. Available: http://www.geneclinics.org/profiles/sca14 เก็บถาวร 2008-05-18 ที่ เวย์แบ็กแมชชีน. (6 May 2008).
แหล่งข้อมูลอื่น[แก้]
| การจำแนกโรค |
|---|
- http://www.ataxia.org(อังกฤษ)
- Cerebellar Degenerations at tchain.com เก็บถาวร 2010-05-27 ที่ เวย์แบ็กแมชชีน(อังกฤษ)
- http://leedsdna.info/tests/DRPLA.htm เก็บถาวร 2011-08-19 ที่ เวย์แบ็กแมชชีน (อังกฤษ)
- http://www.ataxiaforums.co.uk เก็บถาวร 2018-11-04 ที่ เวย์แบ็กแมชชีน
- CureAtaxia.org เก็บถาวร 2011-01-30 ที่ เวย์แบ็กแมชชีน(อังกฤษ)